在现有的仪器分析实验教学平台上,X-射线衍射仪、核磁共振谱仪、气质联用仪、毛细管电泳仪、凝胶渗透色谱仪、气相色谱仪、电感耦合等离子体原子发射光谱仪、原子吸收光谱仪、示差扫描量热计、热重-差热热分析仪、紫外-可见分光光度计、分子荧光分光光度计、傅里叶变换红外光谱仪等十多种仪器设备,已用于“仪器分析实验”课程的教学(涉及至少12种仪器,开设12个实验项目,60学时)以及学生探究性、自主设计性实验,涵盖基础型、综合性和研究型实验三个教学层次。但还是由于大型仪器操作的高度自动化、内部器件又大都被封闭,而且还受到仪器台套数和实际教学学时限制,学生在每一个短短的教学单元内很难做到熟练操作仪器、并深刻理解仪器工作原理。更有一些高成本的大型仪器(如,液相色谱-质谱联用仪、扫描电镜、透射电镜、电感耦合等离子体光谱-质谱联用仪等),目前尚不能用于开设教学实验。
为此,对于一些高成本的、目前尚不能用于开设教学实验的大型仪器设备,构建仿真的虚拟仪器分析实验教学环境,学生通过人机交互完成一个教学实验过程的各个环节,包括样品前处理、仪器参数的设置、整个测试过程的操作、数据处理等,从而对这些大型仪器的工作原理及操作规范有个基本了解。目前我们建有液相色谱-质谱联用仪、电感耦合等离子体光谱-质谱联用仪、扫描电镜、透射电镜、气相色谱-红外光谱联用仪等虚拟仿真实验项目。这些是对真实实验项目的重要补充。
此外,一些已用于开设教学实验的仪器,还可以通过虚拟仿真技术来进一步扩展其教学内容。如现有的“仪器分析实验”课程中色谱类实验的开设是把气相色谱、液相色谱及毛细管电泳合为色谱类综合实验项目,内容是选择合适的色谱分析方法分离并检测出奶茶、雪碧、可乐、芬达饮料中的不同成分,分别是(1)气相色谱仪分离并测定奶茶中胆固醇的含量;(2)液相色谱仪分离并测定奶茶、可乐中咖啡因的含量;(3)毛细管电泳仪分离并测定雪碧、芬达中苯甲酸钠的含量。每位学生可以使用到这三种典型色谱分析方法,通过三个教学单元依次完成三个实验,这对初步了解三种方法的异同点和各自特点有着很大帮助。然而,仪器的应用领域是广泛的,受实际教学学时限制,学生很难做到在一个教学单元里就能深刻理解仪器工作原理与广泛的应用。同样我们可以构建仿真的虚拟仪器分析实验教学环境,拓宽仪器的应用范围,如构建了虚拟仿真实验“毛细管电泳法测定牛奶中的三聚氰胺”,对毛细管电泳法的基本原理,如电泳现象、电渗流现象、淌度,以及毛细管电泳如何利用这些现象将不同离子分开进行了动态展示,还有毛细管电泳仪的工作过程也以动画形式生动形象地演示出来,以进一步加深和强化学生对这些原理的理解。特别是设计了人机交互式样品预处理环节,使学生对这一重要过程有个大致了解,此外,毛细管电泳工作站操作复杂,在虚拟的仪器分析环节,学生可以对每个步骤进行仿真操作,最后,学生通过整理相关实验数据,完成并提交实验报告。这些是对真实实验内容的重要扩展。
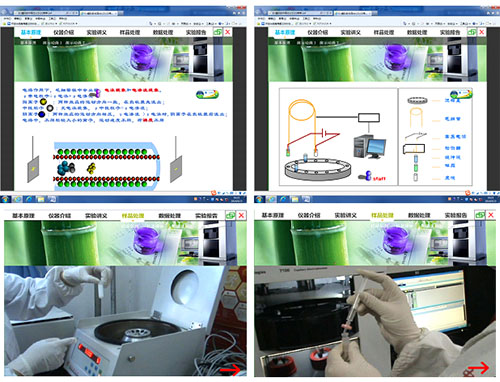
㈠ 现有的实验教学内容
现有的“仪器分析实验”课程中色谱类实验的开设是把气相色谱、液相色谱及毛细管电泳合为色谱类综合实验项目,内容是选择合适的色谱分析方法分离并检测出奶茶、雪碧、可乐、芬达饮料中的不同成分,分别是(1)气相色谱仪分离并测定奶茶中胆固醇的含量;(2)液相色谱仪分离并测定奶茶、可乐中咖啡因的含量;(3)毛细管电泳仪分离并测定雪碧、芬达中苯甲酸钠的含量。每位学生可以使用到这三种典型色谱分析方法,通过三个教学单元依次完成三个实验,这对初步了解三种方法的异同点和各自特点有着很大帮助。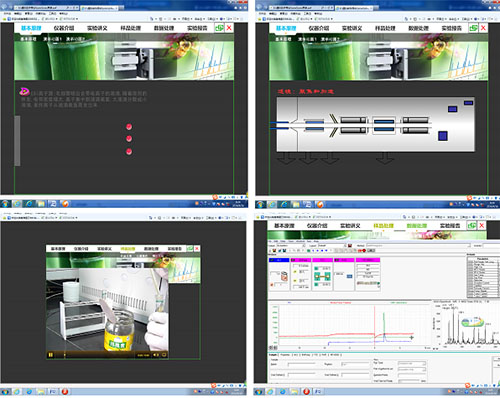
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,拓宽仪器的应用范围。对毛细管电泳法的基本原理,如电泳现象、电渗流现象、淌度,以及毛细管电泳如何利用这些现象将不同离子分开进行了动态展示,还有毛细管电泳仪的工作过程也以动画形式生动形象地演示出来,以进一步加深和强化学生对这些原理的理解。特别是设计了人机交互式样品预处理环节,使学生对这一重要过程有个大致了解,此外,毛细管电泳工作站操作复杂,在虚拟的仪器分析环节,学生可以对每个步骤进行仿真操作,最后,学生通过整理相关实验数据,完成并提交实验报告。
这些是对真实实验内容的重要扩展。有效地解决了受实际教学学时限制,学生在短短的教学单元内很难做到熟练操作仪器、并深刻理解仪器工作原理与应用等问题。
㈠ 现有的实验教学内容
现有的“仪器分析实验”课程没有相关教学内容,这是由于仪器的较高成本、并受台套数限制(一套),目前尚不能用于开设教学实验。
㈡ 虚拟仿真实验内容、功能及效果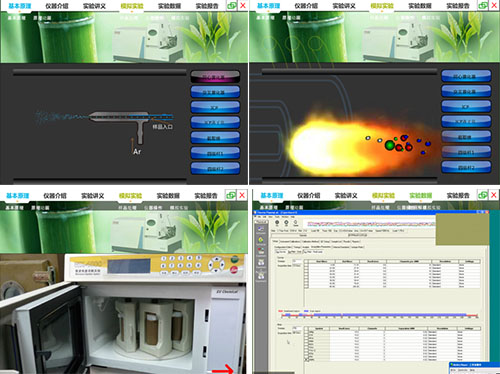
采用虚拟仿真实验的形式,让学生对液质联用仪的基本原理(仪器结构、各种不同的扫描方式、定性定量方法、样品处理以及工作站操作思路等)有个基本了解。模拟电喷雾离子源(ESI)离子化机理、三重四级杆质谱仪的工作原理以及试样的进样前处理过程和工作站操作,并要求学生根据所产生的实验数据完成并提交实验报告。学习并完成本虚拟仿真实验项目,对学生完整了解液质联用仪的基本原理和使用方法无疑是很有帮助的。
本虚拟仿真实验是依照Agilent 6460三重四级杆液相色谱-质谱联用仪来设计的。
㈠ 现有的实验教学内容
由于仪器的高成本,现有的“仪器分析实验”课程没有相关教学内容。
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,模拟ICP-MS基本工作原理,如,雾化器雾化过程、ICP工作过程及离子化机理等,模拟试样的进样前处理过程及工作站操作,并要求学生根据所产生的实验数据提交实验报告,使学生对液质联用仪的基本原理和使用方法有一个完整的了解。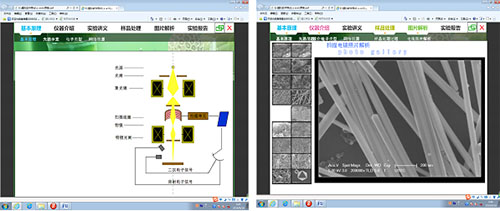
本虚拟仿真实验是依照Thermo X Series 2 ICP-MS来设计的。
㈠ 现有的实验教学内容
由于扫描电子显微镜的价格昂贵、操作复杂、使用耗时等特点,现有的“仪器分析实验”课程尚没有相关教学内容。
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,模拟扫描电子显微镜(SEM)基本工作原理,模拟仪器的基本结构及工作过程,以帮助学生对仪器工作原理有个基本理解,设计了人机交互式样品预处理环节,使学生对样品预处理过程有大致了解,由于扫描电镜操作复杂,在仪器分析环节,本实验为学生设计并模拟了每一步的操作过程。最后根据模拟的电镜图片,对学生进行简单的图片解析训练,并要求学生完成并提交实验报告。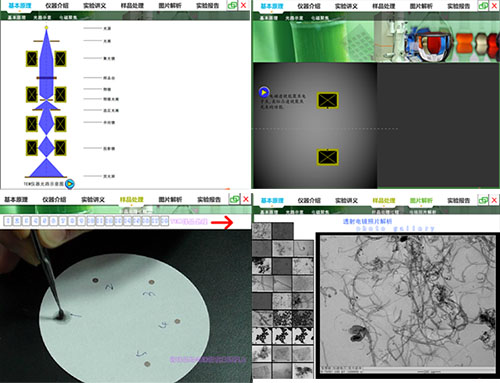
本虚拟仿真实验是依照JSM-6700F扫描电子显微镜来设计的。
㈠ 现有的实验教学内容
同样是由于透射电子显微镜的价格昂贵、操作复杂、使用耗时等特点,现有的“仪器分析实验”课程尚没有相关教学内容。
㈡ 虚拟仿真实验内容、功能及效果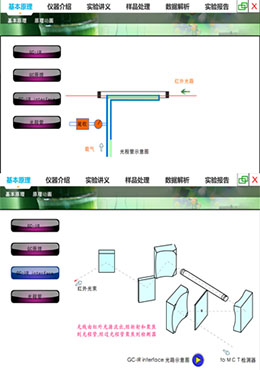
构建仿真的虚拟仪器分析实验教学环境,模拟透射电子显微镜(TEM)基本工作原理,如,聚光镜对电子的聚焦作用、电子束的路径以及最终成像过程等,设计了人机交互式样品预处理环节,以模拟试样的进样前处理过程,对工作站操作也进行了仿真模拟,最后要求学生根据电镜图片,完成图片解析训练,并提交实验报告,使学生对透射电镜的整个操作及测定过程有一个完整的了解。
本虚拟仿真实验是依照Hitachi-7650透射电子显微镜来设计的。
㈠ 现有的实验教学内容
气相色谱-红外联用作为一种新型联用技术,学生对此了解很少,现有的“仪器分析实验”课程也没有相关教学内容。
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,模拟气相色谱-红外联用仪(GC-IR)的基本工作原理,包括,气相色谱的基本原理、红外检测器的基本结构及光路图、光程管的工作过程以及气相色谱红外联用仪的主要结构等,利用虚拟仿真系统进行基本的操作练习,测定简单化合物的红外谱图。此外,通过模拟训练,掌握试样的前处理、工作站操作以及基本的定性定量方法。
本虚拟仿真实验是依照PE-2000气相色谱-红外联用仪来设计的。
为此,对于一些高成本的、目前尚不能用于开设教学实验的大型仪器设备,构建仿真的虚拟仪器分析实验教学环境,学生通过人机交互完成一个教学实验过程的各个环节,包括样品前处理、仪器参数的设置、整个测试过程的操作、数据处理等,从而对这些大型仪器的工作原理及操作规范有个基本了解。目前我们建有液相色谱-质谱联用仪、电感耦合等离子体光谱-质谱联用仪、扫描电镜、透射电镜、气相色谱-红外光谱联用仪等虚拟仿真实验项目。这些是对真实实验项目的重要补充。
此外,一些已用于开设教学实验的仪器,还可以通过虚拟仿真技术来进一步扩展其教学内容。如现有的“仪器分析实验”课程中色谱类实验的开设是把气相色谱、液相色谱及毛细管电泳合为色谱类综合实验项目,内容是选择合适的色谱分析方法分离并检测出奶茶、雪碧、可乐、芬达饮料中的不同成分,分别是(1)气相色谱仪分离并测定奶茶中胆固醇的含量;(2)液相色谱仪分离并测定奶茶、可乐中咖啡因的含量;(3)毛细管电泳仪分离并测定雪碧、芬达中苯甲酸钠的含量。每位学生可以使用到这三种典型色谱分析方法,通过三个教学单元依次完成三个实验,这对初步了解三种方法的异同点和各自特点有着很大帮助。然而,仪器的应用领域是广泛的,受实际教学学时限制,学生很难做到在一个教学单元里就能深刻理解仪器工作原理与广泛的应用。同样我们可以构建仿真的虚拟仪器分析实验教学环境,拓宽仪器的应用范围,如构建了虚拟仿真实验“毛细管电泳法测定牛奶中的三聚氰胺”,对毛细管电泳法的基本原理,如电泳现象、电渗流现象、淌度,以及毛细管电泳如何利用这些现象将不同离子分开进行了动态展示,还有毛细管电泳仪的工作过程也以动画形式生动形象地演示出来,以进一步加深和强化学生对这些原理的理解。特别是设计了人机交互式样品预处理环节,使学生对这一重要过程有个大致了解,此外,毛细管电泳工作站操作复杂,在虚拟的仪器分析环节,学生可以对每个步骤进行仿真操作,最后,学生通过整理相关实验数据,完成并提交实验报告。这些是对真实实验内容的重要扩展。
项目1 毛细管电泳法测定牛奶中的三聚氰胺
㈠ 现有的实验教学内容
现有的“仪器分析实验”课程中色谱类实验的开设是把气相色谱、液相色谱及毛细管电泳合为色谱类综合实验项目,内容是选择合适的色谱分析方法分离并检测出奶茶、雪碧、可乐、芬达饮料中的不同成分,分别是(1)气相色谱仪分离并测定奶茶中胆固醇的含量;(2)液相色谱仪分离并测定奶茶、可乐中咖啡因的含量;(3)毛细管电泳仪分离并测定雪碧、芬达中苯甲酸钠的含量。每位学生可以使用到这三种典型色谱分析方法,通过三个教学单元依次完成三个实验,这对初步了解三种方法的异同点和各自特点有着很大帮助。
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,拓宽仪器的应用范围。对毛细管电泳法的基本原理,如电泳现象、电渗流现象、淌度,以及毛细管电泳如何利用这些现象将不同离子分开进行了动态展示,还有毛细管电泳仪的工作过程也以动画形式生动形象地演示出来,以进一步加深和强化学生对这些原理的理解。特别是设计了人机交互式样品预处理环节,使学生对这一重要过程有个大致了解,此外,毛细管电泳工作站操作复杂,在虚拟的仪器分析环节,学生可以对每个步骤进行仿真操作,最后,学生通过整理相关实验数据,完成并提交实验报告。
这些是对真实实验内容的重要扩展。有效地解决了受实际教学学时限制,学生在短短的教学单元内很难做到熟练操作仪器、并深刻理解仪器工作原理与应用等问题。
项目2 液质联用法测定蜂蜜中的氯霉素
㈠ 现有的实验教学内容
现有的“仪器分析实验”课程没有相关教学内容,这是由于仪器的较高成本、并受台套数限制(一套),目前尚不能用于开设教学实验。
㈡ 虚拟仿真实验内容、功能及效果
采用虚拟仿真实验的形式,让学生对液质联用仪的基本原理(仪器结构、各种不同的扫描方式、定性定量方法、样品处理以及工作站操作思路等)有个基本了解。模拟电喷雾离子源(ESI)离子化机理、三重四级杆质谱仪的工作原理以及试样的进样前处理过程和工作站操作,并要求学生根据所产生的实验数据完成并提交实验报告。学习并完成本虚拟仿真实验项目,对学生完整了解液质联用仪的基本原理和使用方法无疑是很有帮助的。
本虚拟仿真实验是依照Agilent 6460三重四级杆液相色谱-质谱联用仪来设计的。
项目3 电感耦合等离子体-质谱法测定茶叶中的微量元素
㈠ 现有的实验教学内容
由于仪器的高成本,现有的“仪器分析实验”课程没有相关教学内容。
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,模拟ICP-MS基本工作原理,如,雾化器雾化过程、ICP工作过程及离子化机理等,模拟试样的进样前处理过程及工作站操作,并要求学生根据所产生的实验数据提交实验报告,使学生对液质联用仪的基本原理和使用方法有一个完整的了解。
本虚拟仿真实验是依照Thermo X Series 2 ICP-MS来设计的。
项目4 扫描电子显微镜
㈠ 现有的实验教学内容
由于扫描电子显微镜的价格昂贵、操作复杂、使用耗时等特点,现有的“仪器分析实验”课程尚没有相关教学内容。
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,模拟扫描电子显微镜(SEM)基本工作原理,模拟仪器的基本结构及工作过程,以帮助学生对仪器工作原理有个基本理解,设计了人机交互式样品预处理环节,使学生对样品预处理过程有大致了解,由于扫描电镜操作复杂,在仪器分析环节,本实验为学生设计并模拟了每一步的操作过程。最后根据模拟的电镜图片,对学生进行简单的图片解析训练,并要求学生完成并提交实验报告。
本虚拟仿真实验是依照JSM-6700F扫描电子显微镜来设计的。
项目5 透射电子显微镜
㈠ 现有的实验教学内容
同样是由于透射电子显微镜的价格昂贵、操作复杂、使用耗时等特点,现有的“仪器分析实验”课程尚没有相关教学内容。
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,模拟透射电子显微镜(TEM)基本工作原理,如,聚光镜对电子的聚焦作用、电子束的路径以及最终成像过程等,设计了人机交互式样品预处理环节,以模拟试样的进样前处理过程,对工作站操作也进行了仿真模拟,最后要求学生根据电镜图片,完成图片解析训练,并提交实验报告,使学生对透射电镜的整个操作及测定过程有一个完整的了解。
本虚拟仿真实验是依照Hitachi-7650透射电子显微镜来设计的。
项目6 气相色谱-红外光谱联用仪
㈠ 现有的实验教学内容
气相色谱-红外联用作为一种新型联用技术,学生对此了解很少,现有的“仪器分析实验”课程也没有相关教学内容。
㈡ 虚拟仿真实验内容、功能及效果
构建仿真的虚拟仪器分析实验教学环境,模拟气相色谱-红外联用仪(GC-IR)的基本工作原理,包括,气相色谱的基本原理、红外检测器的基本结构及光路图、光程管的工作过程以及气相色谱红外联用仪的主要结构等,利用虚拟仿真系统进行基本的操作练习,测定简单化合物的红外谱图。此外,通过模拟训练,掌握试样的前处理、工作站操作以及基本的定性定量方法。
本虚拟仿真实验是依照PE-2000气相色谱-红外联用仪来设计的。